Bin
Bin
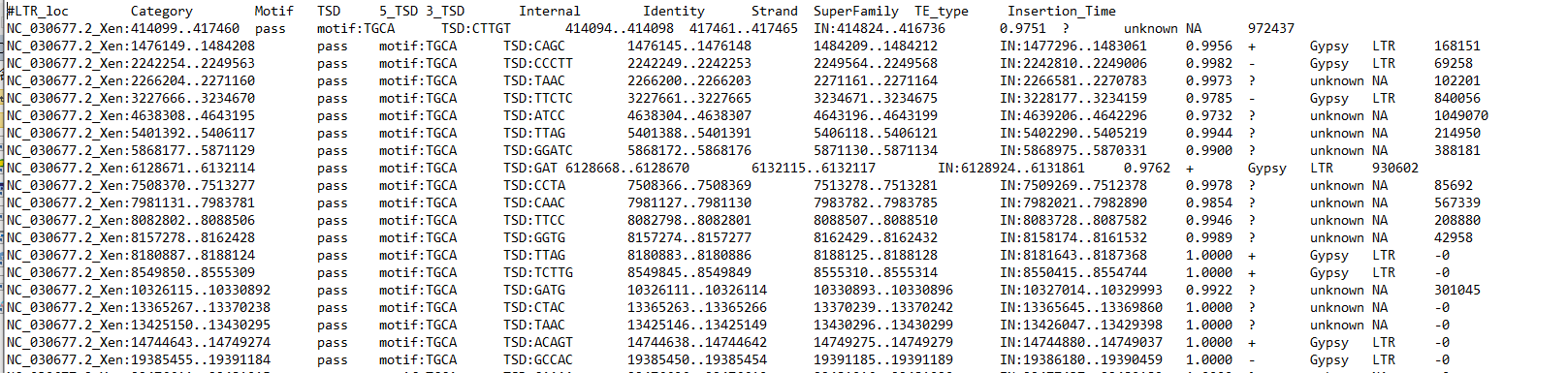
I have one more question for you. What do I do if I have many genomes to study, but the natural mutation rate is known for only one species? Can...
I have received your email. Thank you for your reply ------------------ 原始邮件 ------------------ 发件人: "oushujun/LTR_retriever" ***@***.***>; 发送时间: 2022年8月22日(星期一) 上午10:45 ***@***.***>; ***@***.******@***.***>; 主题: Re: [oushujun/LTR_retriever] LTR insertion time (Issue #130) Sorry somehow I didn't send...
Dear author, I currently have data sets of HIFI and HIC. Is the assembly result of HIC and HIFI input together better than that of HIFI alone? I need the...
Thank you. I think so, too. But I don't know if there is any difference in the assembly quality of the genome as a result of using the two methods发自我的 ...
Thank you very much for your answer, but I am curious about how you can assemble X and Y chromosomes like humans. I currently have HiFI and HIC data发自我的 iPhone在...
Yes,this is my code. # 数据预处理 liger_obj % normalize() %>% selectGenes() %>% scaleNotCenter() # 使用runIntegration进行矩阵整合 liger_obj
Thanks, I will try this method All best!! ------------------ 原始邮件 ------------------ 发件人: "welch-lab/liger" ***@***.***>; 发送时间: 2025年4月17日(星期四) 晚上9:41 ***@***.***>; ***@***.******@***.***>; 主题: Re: [welch-lab/liger] Cross-species integrated annotation (Issue #328) Thanks, Please try replacing quantileNorm() with centroidAlign(),...
Dear Wang, I have attempted to replace quantileNorm() with centroidAlign() as an alternative approach. However, the results show that the same cell types across multiple species are not properly integrated...