termithorbor
termithorbor
I tried it but it works not as good as I expected it So after running cutadapt my first primer pair is still found in some places: Before: 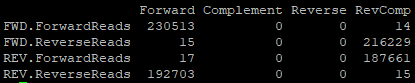 After:...
I tryed it and now make works but when I want to run dashing my system says that it can not find the command dashing. However I am in the...
I had the error as well. Reinstalling Masurca with Conda worked for me. https://anaconda.org/bioconda/masurca
The goal is just to get rid of taxa which might be artefacts as mentioned in the paper. So is such a filtering approach reasonable or is it not needed...
Did you use Conda for installing Unicycler? if so you can run conda update spades or conda install spades=3.15.5
Is it normal, that the command runs for several days on 12 paired and samples with fastq files around 12 Gb for each forward and reverse reads on a server...
So far only empty gene_quant folders but no further data was produced. In the terminal no timestaps/intermediate steps are shown:  It is already running for ~5 days.
I am running version 2.0.4: watch -n 5 free -m Every 5,0s: free -m dil-sequenz: Wed Apr 10 13:55:36 2024 total used free shared buff/cache available Mem: 806288 16460 67087...
Okay, however it is already running for 8 days. Is this still normal behaviour?
Still running...I fear it is stuck somewhere I have restarted it now with the --no_count_split_by_alignment_no option. Or is using --no_count_splitting_by_gene_no as well a better option?