SPADES changed flag for single-read in illumina reads
PR checklist
Changed flag for single-read illumina reads to --s, since -s is used in spades for singletons from a pair-end library (see Input data in https://github.com/ablab/spades/blob/spades_3.15.5/README.md)
- [x] This comment contains a description of changes (with reason).
- [ ] If you've fixed a bug or added code that should be tested, add tests!
- [ ] If you've added a new tool - have you followed the module conventions in the contribution docs
- [ ] If necessary, include test data in your PR.
- [ ] Remove all TODO statements.
- [x] Emit the
versions.ymlfile. - [ ] Follow the naming conventions.
- [ ] Follow the parameters requirements.
- [ ] Follow the input/output options guidelines.
- [ ] Add a resource
label - [x] Use BioConda and BioContainers if possible to fulfil software requirements.
- Ensure that the test works with either Docker / Singularity. Conda CI tests can be quite flaky:
- [x]
PROFILE=docker pytest --tag <MODULE> --symlink --keep-workflow-wd --git-aware - [ ]
PROFILE=singularity pytest --tag <MODULE> --symlink --keep-workflow-wd --git-aware - [ ]
PROFILE=conda pytest --tag <MODULE> --symlink --keep-workflow-wd --git-aware
- [x]
Is this actually correct?
--scomes under the 'multiple libraries' section, where my understanding of the implementation of this module is that it only ever accepts a single library (i.e.-1 vs -2, rather than--pe-1 -0pe-2 etc.?The docs below would imply to me that for single-end libraries you would just give
-1?
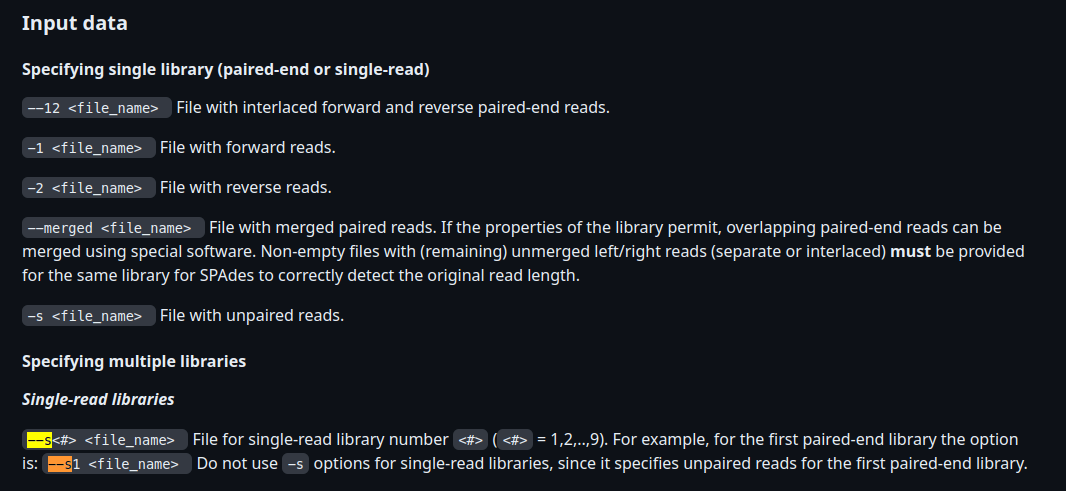
I tried to add the -1 flag as suggested by @jfy133, however when I used the -1 flag for the single-end alone the following error occurs:
== Error == the number of files with left paired reads is not equal to the number of
files with right paired reads (library number: 1, library type: paired-end)!
When I used the `--s1`` the input is interpreted correctly:
Dataset parameters:
RNA virus assembly mode
Metagenomic mode
Reads:
Library number: 1, library type: single
left reads: not specified
right reads: not specified
interlaced reads: not specified
single reads: ['/private/var/folders/1c/72tb52hs181_9xn2l2s9xt4m0000gp/T/tmp51itgo6g/63/1b394bce5e84e0aff90f28e1af303c/test_2.fastq.gz']
merged reads: not specified
But I agree that the documentation seems to indicate otherwise
Hi @SPPearce, I just returned from maternity leave, I will try to get to this within the next month.
@aidaanva We are doing some nf-core cleaning and trying to close PRs when not used anymore (they can be reopened in the future ofc). Just to know, are you planning to work on this one? :)
I donor have the time to work on this one right now. I will reopen it when I have the time. Thank you for asking.