Suggested edits to enable linking to CoVariants pages, others in the future
This PR generates an extra annotation in the tooltips after the PCR Primer changes, listing the CoVariants page(s) that correspond to the given sequence. The linking is done via variant constellation definitions in constellations.json
This pull request is being automatically deployed with Vercel (learn more).
To see the status of your deployment, click below or on the icon next to each commit.
🔍 Inspect: https://vercel.com/nextstrain/nextclade/4bCKM8964os9mcw6r9HMYyRMvKUS
✅ Preview: https://nextclade-git-fork-nodrogluap-constellations-nextstrain.vercel.app
Added existing TypeScript file changes to the branch, initial PR just included the 5 new files.
Code Climate warning not something that's easy to fix without refactoring the original code, as the whole function is structured to do a bunch of returns? My code commit did not add any return statements.
Hi @nodrogluap,
Nice work, thanks!
I mostly handle the dev part and I'll let @rneher and @emmahodcroft to review the scientific part. I might be out of context here, but here is a few thoughts/questions:
-
Could you please provide a bit more scientific background about these changes? A particularly confusing part for me is clades vs variants. Looks like the current situation in the community is a bit of a mess.
-
Where does the data in
constellations.jsoncome from? How it should be updated over time? Should it be synchronized with covariants.org? -
Do you see any problems that may arise long-term?
-
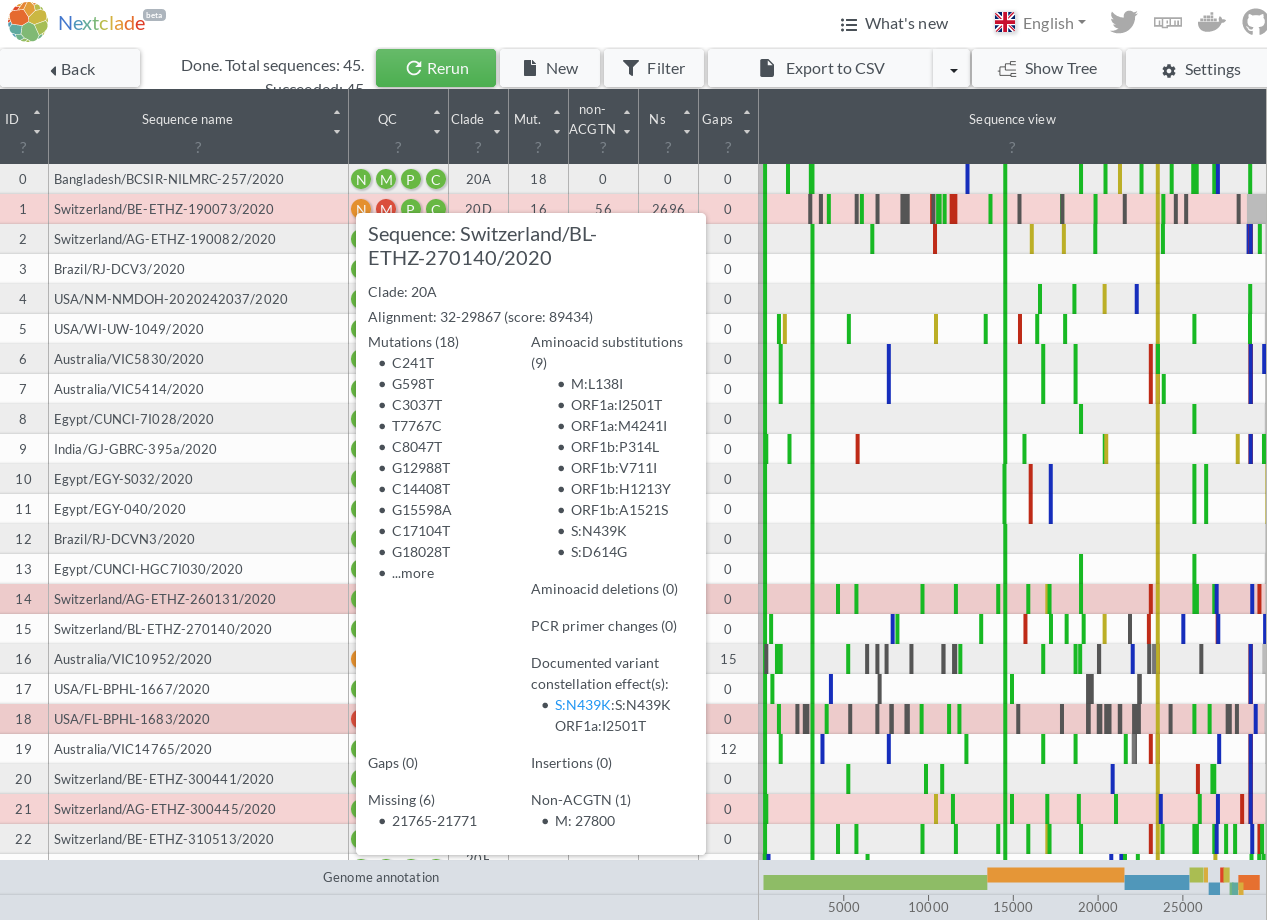
I am a bit confused as to what are all the things shown after the variant link? Unless this is a widespread notation, maybe we could present this information in a structured manner?
-
Should we also export this information into CSV/JSON outputs (for both, web and CLI versions)?
-
It's a bit challenging to click on links in the tooltip, as the tooltips tend to disappear on mouse move. Might be a good reason to finally implement #83.
-
Ignore codeclimate. I found that for a small project such as Nextclade its warnings don't provide a lot of value. We might remove it.
Hi @nodrogluap ! Thanks so much for making a PR on this! I still think this idea is a great one, but it's going to take me a little bit until I can pick through the edits - but definitely will take a good look!
Could you please provide a bit more scientific background about these changes? A particularly confusing part for me is clades vs variants. Looks like the current situation in the community is a bit of a mess.
Great question :-) I'm not married to the wording before the link, I've (mal?)adopted Andrew Rambaut's 'constellation' terminology to describe a set of variants that do not necessarily arise from the same tree branch, yet are showing up and probably mean something biologically. Note that SNP 'cluster' already has a different meaning in the NextClade QC UI, so constellation seemed like the thing to call it.
Where does the data in constellations.json come from? How it should be updated over time? Should it be synchronized with covariants.org?
With this initial commit I've extracted the minimal information from CoVariant's clusters.json file that are required to define the link out: the defining variant(s) and a name/description, and I've added the URL. In theory you could add a method to convertConstellationDefinitions.ts to directly accept the clusters.json file, but I'd like to keep the generic minimal JSON input function too, as part of my motivation is the ability to re-use the NextStrain UI with a custom link out dataset for my own purposes.
I am a bit confused as to what are all the things shown after the variant link? Unless this is a widespread notation, maybe we could present this information in a structured manner?
The list after the link is the set of variants that define the constellation matched. e.g. When the constellation matched is L452R, this list of variants required to be matched for the CoVariant information is actually three spike variants and two ORF1 variants (S:S13I S:W152C S:L452R ORF1a:I4205V ORF1b:D1183Y). This display separates the logic of the variant matching from the naming scheme. Once again, I have a selfish motivation, because if I upload a custom JSON, I might want a link that says "possibly anthropozoonotic" and list the one variant in the analyzed genome that has that property such as Y453F.
Should we also export this information into CSV/JSON outputs (for both, web and CLI versions)?
That's included in the commit.
but it's going to take me a little bit until I can pick through the edits - but definitely will take a good look!
Indeed, adding this one bit to the tooltips required 5 new files and modifying 22 existing files with all the triggers, callbacks, defaults loading, etc. :-) In essence, I adapted the existing code structure for the PCR Primer Changes list that precedes it in the UI.
I'm just starting to pick into this PR - sorry for the delay! So I think the linking out to CoVariants is great and useful. I'm hoping that now that many of the variants & mutations are split out, this could be easier - one can link to a variant if it matches that description, or just to a mutation if it has a 'mutation of interest' that people may want more information about.
I'm hesitant to get into 'constellations' because I don't really track these on CoVariants - everything is either a variant (which is a monophyletic cluster) or mutation. I recognise this is a recent change, though!
I am hoping to break out a few more mutations in CoVariants so that linking to mutation pages is easy and straightforward. That part should hopefully be pretty hassle-free - we can read in a list of anything that's a 'mutation' from the CoVariants clusters.py file (or the generated json file), and just link to the page.
For Variants it might be a little harder - I notice above some discussion about how many mutations should match to call it or not. I feel like some mutations may be more important than others - though going by mutations alone is never going to be perfect without taking into account the phylogeny. We could possibly define a 'key mutations' list that is required to 'match' which might be a subset of the 'full' mutation list.
@ivan-aksamentov one option that may make pulling things from CoVariants to NextClade easier is we could break out the 'mutations' sections of clusters.py if you like - so that these can be accessed separately? (I don't know if that's easier or not!) Possibly then it could be used both for NextClade and for the 'Mutation Comparison' part of CoVariants.
Definitely agree that the default linking out JSON in NextClade should be carefully considered vis-a-vis how you intend CoVariants to grow. While the added source files include the word "constellation" (since "variant cluster" is already used in NextClade), there is no constraint on the meaning of "constellation" in the code. The key variants idea for monophyletic definition is a good one I think!
This PR basically creates a JSON mechanism to link out from a set of variants to a Web page. We are starting to use this feature at our public health lab, and selfishly I'd like to have that mechanism in the main branch so I don't need to remerge my fork whenever NextClade gets updated. :-). I'm more than happy with whatever JSON definitions and UI wording you feel should be include as the defaults, we would be supplementing with a custom JSON file anyway internally...
@nodrogluap @emmahodcroft Happy to proceed. I think we might:
- make a script to generate the
constellations.jsonin Covariants repo fromclusters.py. Please do :) as I am not qualified. Should be pretty much the addition of 2 files. If data can be derived as is, I don't think any modifications toclusters.pyare needed. - Emma will then run this script as a part of the usual data update routine
- Setup a bot to pull-request
constellations.jsoninto Nextclade, periodically or with a webhook. Optionally with a manual trigger. - Use this code (I mean in this PR) to render the JSON in Nextclade
- Optionally, prettify how the mutations are listed, because right now it's often a large blob of characters
Thoughts?