mouse CT26 samples with strange copy number
This is my first analysis and I ran FACETS on CT26 mouse tumors and Normal tissues (taken from literature). I obtain strange results because all the segments seems to be with 1 copy number (i expected some segments with diploid state).
Please could you give your opinion about the correctness of the results that I obtained?

This is the code that I used set.seed(1234)set.seed(1234) rcmat<-readSnpMatrix(filename = "tableSNP") xx<-preProcSample(rcmat,gbuild = "mm10",unmatched = T,) oo<-procSample(xx,cval=150) fit <-emcncf(oo)
Thank you very much
The difficulty with (lab) mouse data is that unlike humans the proportion of het snps is very small and the het loci are not spread somewhat uniformly. This leads to very poor estimate of dipLogR (log-ratio corresponding to diploid state). You are seeing a result of that. You can try giving an alternate dipLogR (say around the green line in the plot) in procSample and see if that helps.
Venkat
Thank you for you comment. I ran another trial by lowering the dipLogR and changing some parameters library(facets) set.seed(1234) rcmat<-readSnpMatrix(filename = "tableSNP") xx1<-preProcSample(rcmat,gbuild = "mm10",snp.nbhd = 250) oo1<-procSample(xx1,dipLogR = 0,cval=300,min.nhet = 15) fit1 <-emcncf(oo1)
Results are more reasonably but I see now several regions with NA estimates.
Moreover the X chr is with 0 copynumbers.
Lowering the number of SNPs needed for evaluation(min.nhet) and increasing the size of segments (cval) seems to improve the detection but not so much
Please could you give to me any tips about the most reasonable combinations of parameters to try for this problematic case?
Thank you
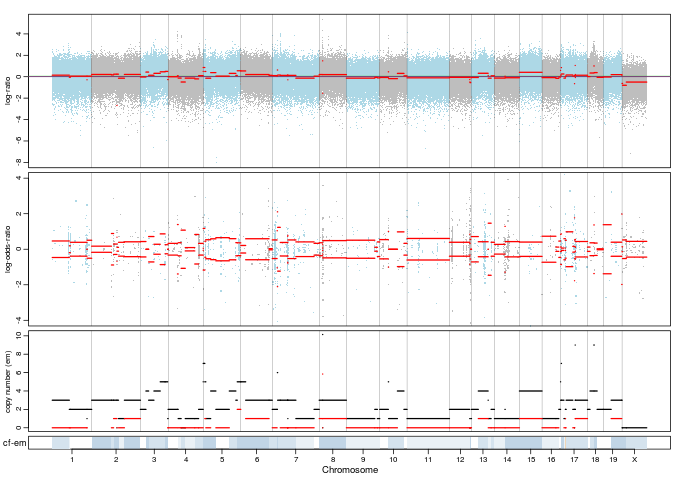
The NA estimate is again because of the nature of the sample. There are very few hets in the entire genome and so you often end up with fewer than min.nhet hets in a segment. Hence the NA. You can try reducing min.nhet further but reliability of any estimate is highly questionable. Unfortunately this is a problem that doesn't have a satisfactory solution (other than have mice with more hets :-)).
Venkat