the number of scaffolds generated by ragtag is not ideal
Hello, Thanks for your application, ragtag. I used Ragtag software to mount my assembled genome, but the reference genome had 16 chromosomes, and the genome I generated could only be mounted to 15. How can I debug to get the correct number of genomes. Looking forward to your reply. Rilla
Hi there,
Could you share your command along with the content of ragtag.scaffolds.stats. Perhaps you could also provide more details about the reference and query assemblies, i.e what species and expected number of chromosomes (sounds like 16 for the reference)?
Thanks
Hi, thanks for your reply. The assembled species is Apis andreniforms and the expected number of chromosomes is 16(16 for reference).The draft genome contain 20 contigs, ande I use the relative species as the guide, below is my command and my ragtag.scaffolds.stats. Thank you!
ragtag.py correct ref.fasta genome_query.fasta -t 5
ragtag.py scaffold ref.fasta genome_query.fasta -t 5
placed_sequences placed_bp unplaced_sequences unplaced_bp gap_bp gap_sequences 17 219153659 3 708984 200 2
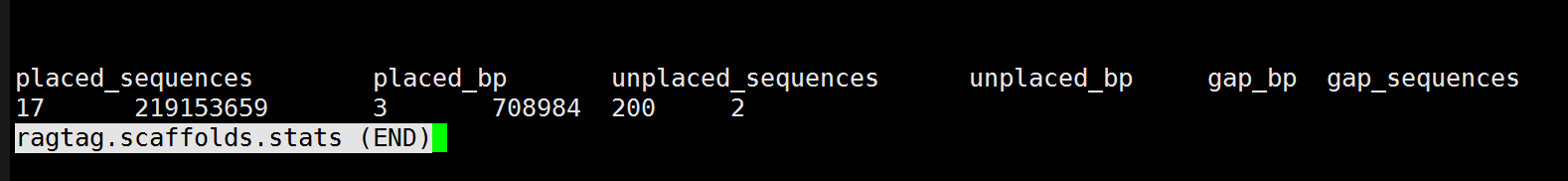
Hi there,
So the vast majority of sequence is being assigned to contigs. One observation is that even though you are using ragtag.py correct, you are not actually using the corrected output as input for scaffolding.
Really the best way to tell what is going on is to make a dotplot with mummerplot or something along those lines. This should tell us if there is a bug in ragtag but just a problem with the query assembly.