Accessing BioCAM .brw files through neo
I'm working on BioCAM MEA recordings, and so far I've been able to use the standalone HerdingSpikes sorter without issues, reading directly from the proprietary .brw files.
However, I'd like to be able to work with SpikeInterface to compare sorters and export outputs as neo spike trains.
The docs mention that spikeinterface.extractors.read_biocam is a "Class for reading data from a Biocam file from 3Brain. Based on neo.rawio.BiocamRawIO". But the current read_biocam function is located in spikeinterface.extractors.neoextractors.biocam, and it seems to be based on probeinterface.read_3brain.
As I've had varying results between standalone and SpikeInterface-based HerdingSpikes, and as I would like to homogenize my workflow as much as possible, is it possible to independently load a Neo recording object and then run HerdingSpikes through SpikeInterface on said object ?
On another, related note : is it possible to access HerdingSpikes Detection and Clustering objects through SpikeInterface somehow ? They have very convenient visualization functions that I would like to access without having to go through spike sporting again with the standalone version.
Hi @BptGrm
I'm working on BioCAM MEA recordings, and so far I've been able to use the standalone HerdingSpikes sorter without issues, reading directly from the proprietary .brw files.
However, I'd like to be able to work with SpikeInterface to compare sorters and export outputs as neo spike trains. The docs mention that
spikeinterface.extractors.read_biocamis a "Class for reading data from a Biocam file from 3Brain. Based on neo.rawio.BiocamRawIO". But the currentread_biocamfunction is located inspikeinterface.extractors.neoextractors.biocam, and it seems to be based onprobeinterface.read_3brain.
Almost all read functions in SI are based on NEO objects. The probeinterface.read_3brain is used just to load the probe information (basically channel locations) that are attached to the spikeinterface object.
As I've had varying results between standalone and SpikeInterface-based HerdingSpikes, and as I would like to homogenize my workflow as much as possible, is it possible to independently load a Neo recording object and then run HerdingSpikes through SpikeInterface on said object ?
No currently it is not possible, but the NEO object maps one to one to the SpikeInterface object so there should be no discrepancies there.
On another, related note : is it possible to access HerdingSpikes Detection and Clustering objects through SpikeInterface somehow ? They have very convenient visualization functions that I would like to access without having to go through spike sporting again with the standalone version.
Currently not, the implemented spike sorters are the full pipelines. However, we are working on modularizing different steps in the sortingcomponents module.
As I've had varying results between standalone and SpikeInterface-based HerdingSpikes, and as I would like to homogenize my workflow as much as possible, is it possible to independently load a Neo recording object and then run HerdingSpikes through SpikeInterface on said object ?
herdingspikes is based on spikeinterface for input.
So if you do
rec = si.read_biocam()
sorting = si.run_sorter('herdingspikes', rec, output_folder='...')
you should have the same result as running directly in HS. So reason why you would not have same results is that default params are not the same.
@mhhennig : what is the best way to run HS ? from SI with run_sorter() or independantly with internal HS machinery ?
@alejoe91 Thank you for the answer !
@samuelgarcia I opened the same file with the two different readers :
recording = read_biocam(data_path) from SI and Probe = BioCam(data_path) from HS,
and then ran HSDetection with the exact same parameters on both.
When displaying the same spike through PlotTracesChannels, I get the following results :
- Through SI :
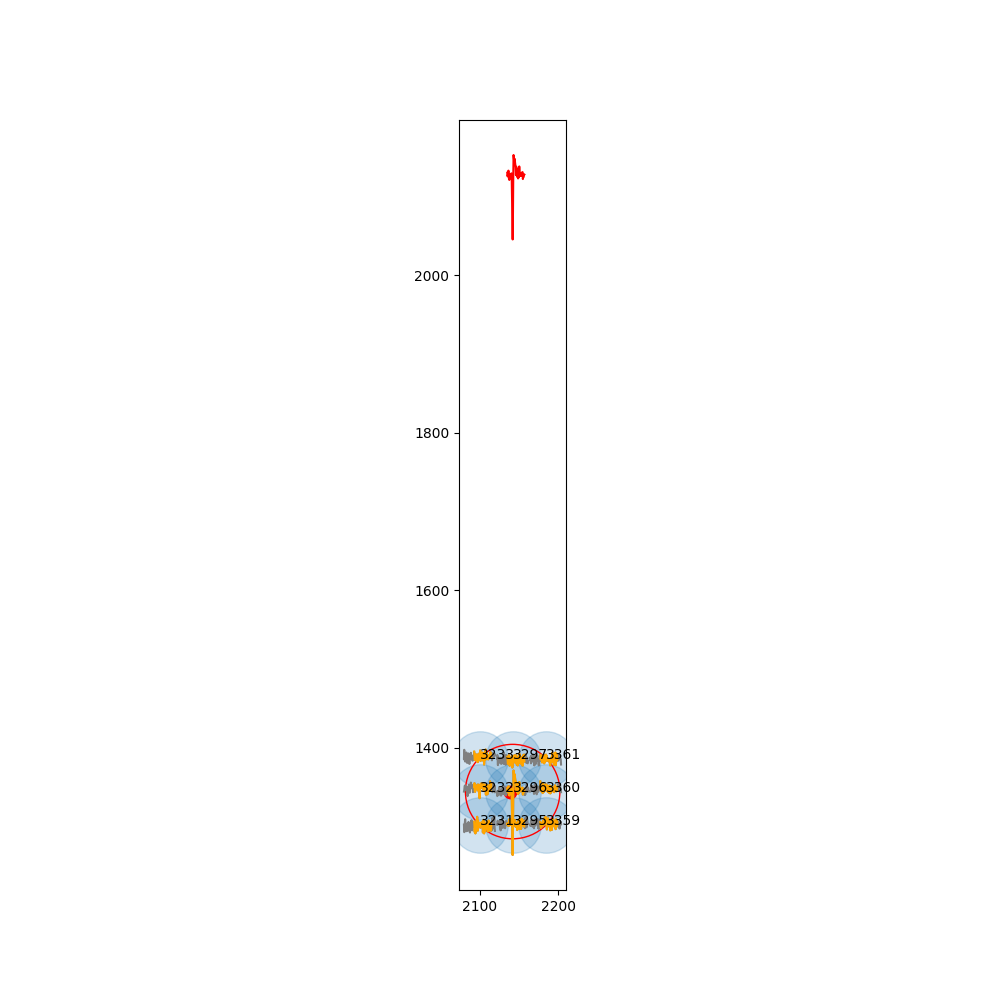
- Through HS :
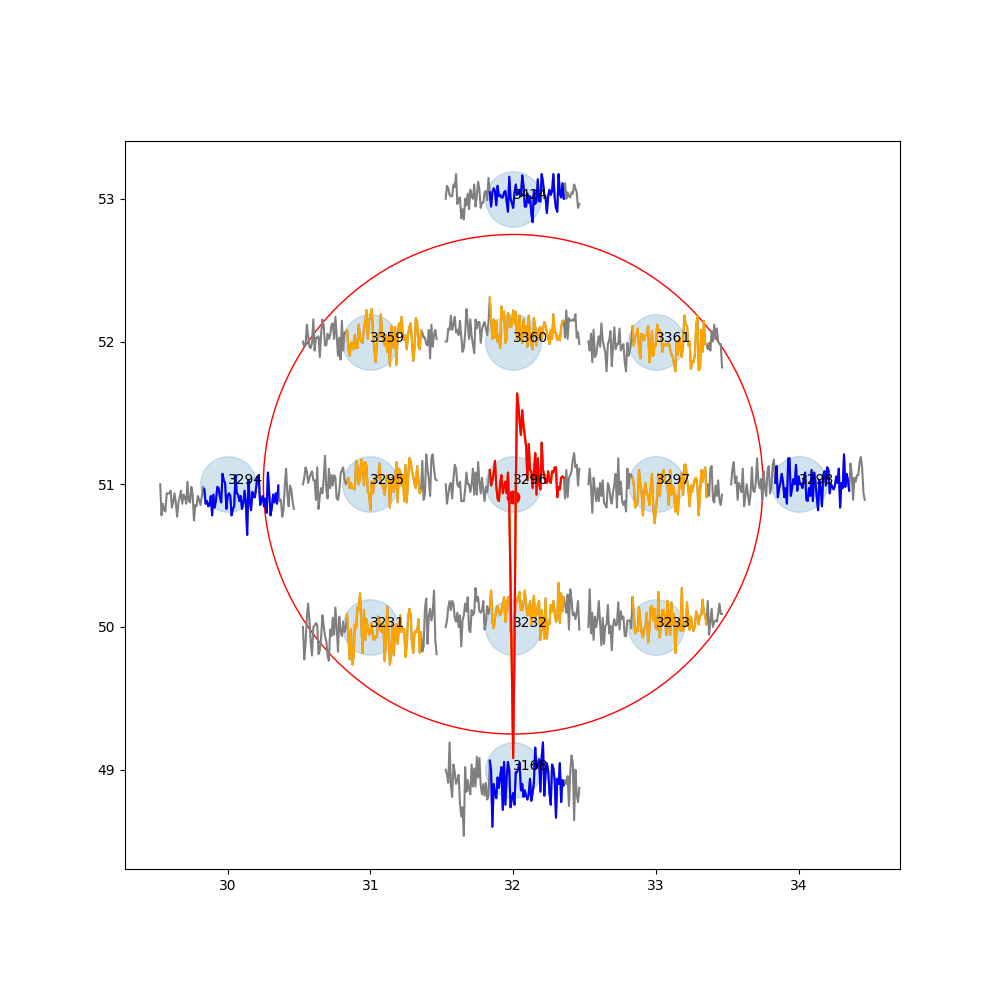
In that case I guess there's something in the preprocessing of si.run_herdingspikes that is necessary ?
I know the localization functions are more specific to HS, I apologize if it's off-topic for a SI issue.
As I understand, so far the best way would be to run HS both standalone to get HS-specific graphs, and as part of SI to get a neo-compatible spike train.
OK I get it.
If the reader make the difference, there is a bug somewhere.
@mhhennig : Any idea why BioCam would be different than read_biocam() ?
@BptGrm : could you try plotting with spikeinterface the channel map ?
sw.plot_probe_map(rec, with_channel_ids=True, with_contact_ids=True)
maybe it is a geometry issue in probeinterface.
maybe it is a geometry issue in probeinterface.
Seems to be ! I had to remove with_contact_ids=True as it was considered an unexpected argument, it seems it is not passed to plot_probe_group properly.
The result is this :
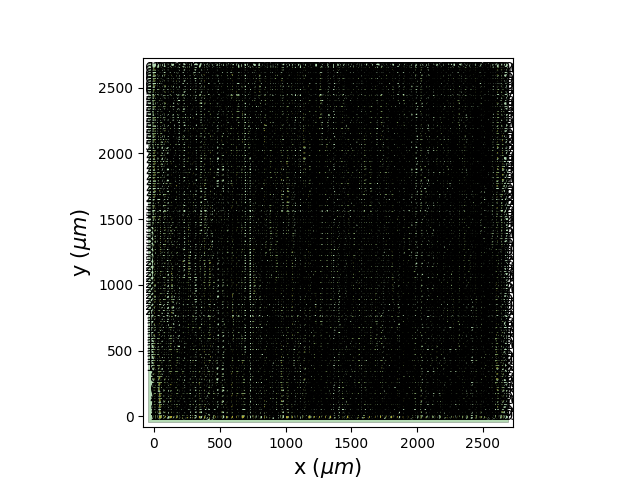 I don't know if electrode pitch is supposed to be passed on in the metadata or if the less than 3mm size is the default plotting scale, but it does not match the size of the arrays recorded.
I don't know if electrode pitch is supposed to be passed on in the metadata or if the less than 3mm size is the default plotting scale, but it does not match the size of the arrays recorded.
When zooming in on the picture I noticed something else :
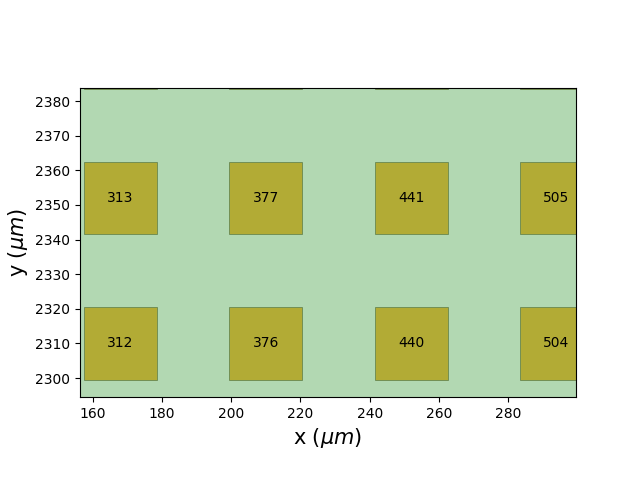 Apparently electrode IDs are incremented in columns going bottom to top then left to right, instead of the usual, in rows left to right then top to bottom.
Apparently electrode IDs are incremented in columns going bottom to top then left to right, instead of the usual, in rows left to right then top to bottom.
Hi,
So the probeinterface implementation is a port from spikeectractors and written by @mhhennig:
You can see that the locations are extracted here (rawIndices) and used as channel locations.
In probeinterface we use the same logic (see here).
HS2 seems to be using some internal file?
Honestly I'm not too familiar with the file format, so probably the best way to double check is to ask someone at 3brain to double check and validate the implementation.
@BptGrm are you in touch with 3Brain? Could you reach out and ask for assistance?
Cheers Alessio
@BptGrm are you in touch with 3Brain? Could you reach out and ask for assistance?
I am, I'll ask them about it !
The intermediate solution I've found so far is to run standalone HerdingSpikes, then pass the output through HerdingspikesSortingExtractor to end up with a SpikeInterface object.
@BptGrm are you in touch with 3Brain? Could you reach out and ask for assistance?
I am, I'll ask them about it !
The intermediate solution I've found so far is to run standalone HerdingSpikes, then pass the output through
HerdingspikesSortingExtractorto end up with a SpikeInterface object.
But then you won't have the recording object available, which is needed for waveforms, locations, etc..
BTW, how did you run the PlotTracesChannels from the SpikeInterface object?
But then you won't have the recording object available, which is needed for waveforms, locations, etc..
That is true, but HerdingSpikes itself can handle at least some of it, and for now I'm mainly looking to extract basic spike train metrics through something like Elephant. Of course having both would be ideal.
BTW, how did you run the
PlotTracesChannelsfrom the SpikeInterface object?
I called it on a HSDetection object, both time. The only difference was the data I fed to HSDetection, i.e. the same file but opened through HS or SI.
How did you instantiate the
HSDetectionfrom a spikeinterface object? Did you use the this class?
I did, I forgot to mention it in the first post. Could that be the issue ?
I dug a bit deeper, and it seems that using RecordingExtractor from HS is indeed causing troubles. The default parameters for inner_radius, neighbor_radius and masked_channels are different, and even when passing them explicitly through Probe = hs.probe.RecordingExtractor(recording,noise_amp_percent=1,inner_radius=1.75,neighbor_radius=None, masked_channels=[0]), the max_neighbors property is different.
As mentionned here :
HS2 seems to be using some internal file?
HS uses internal files for positions_biocam and neighbormatrix_biocam when using the BioCam reader, but they are generated again as positions_spikeextractor and neighbormatrix_spikeextractor when using RecordingExtractor.
I checked and they are indeed very different from each other (see : neighbormatrix.zip)
My guess would be that the issue comes from how ch_positions is obtained in RecordingExtractor, but I haven't been able to find the issue yet.
Please let me know if this is too HS-specific, I can move the issue there !
Yeah maybe open an issue also on the HS page since it's very specific. We'll keep it open also here for the time being :)
@alejoe91 Thanks for the help ! There are also a few runtime warnings I get when using HS through a regular SI pipeline, but I don't know if those are SI-specific or related to the same issue.
For instance, some of them are raised during preprocessing with run_herdingspikes :
C:\conda\envs\electrophy\lib\site-packages\neo\rawio\biocamrawio.py:183: RuntimeWarning: overflow encountered in long_scalars
return rf['3BData/Raw'][nch * t0:nch * t1].reshape((t1 - t0, nch), order='C')
C:\conda\envs\electrophy\lib\site-packages\spikeinterface\toolkit\preprocessing\normalize_scale.py:18: RuntimeWarning: invalid value encountered in multiply
scaled_traces = traces * self.gain[:, channel_indices] + self.offset[:, channel_indices]
Those might be due to metadata being passed improperly, although they only happen with run_herdingspikes and not with standalone HS running on a SI object.
@BptGrm curious if you have an update/workaround for this!
I actually do ! Sorry I forgot to update this issue. I haven't found the actual root cause yet, but it seems that BioCam recordings read through SpikeInterface have a set + 2048 applied to every single data point, e.g. in this case every spike is listed as 2048 µV higher than it actually is.
This explains the problem I had in my second post in this issue, where the y coordinates were simply based on spike voltage.
Oddly enough, this is not a problem if the data is filtered through SI, at least using the bandpass filter I have tested so far.
I usually keep a 300 - 3000 Hz bandpass in my preprocessing now, but I guess the range of frequencies could be extended if someone wants to work on data closer to raw and still work around this.
Accidentally found the culprit in neo while trying to implement BRW v4.x compatibility. In here :
def readHDF5t_100(rf, t0, t1, nch):
return rf['3BData/Raw'][t0:t1]
def readHDF5t_100_i(rf, t0, t1, nch):
return 4096 - rf['3BData/Raw'][t0:t1]
def readHDF5t_101(rf, t0, t1, nch):
return rf['3BData/Raw'][nch * t0:nch * t1].reshape((t1 - t0, nch), order='C')
def readHDF5t_101_i(rf, t0, t1, nch):
return 4096 - rf['3BData/Raw'][nch * t0:nch * t1].reshape((t1 - t0, nch), order='C')
Compared to HerdingSpikes :
def readHDF5t_100(rf, t0, t1, nch):
''' Transposed version for the interpolation method. '''
if t0 <= t1:
d = 2048 - rf['3BData/Raw'][t0:t1].flatten('C').astype(ctypes.c_short)
d[np.where(np.abs(d) > 1500)[0]] = 0
return d
def readHDF5t_100_i(rf, t0, t1, nch):
''' Transposed version for the interpolation method. '''
if t0 <= t1:
d = rf['3BData/Raw'][t0:t1].flatten('C').astype(ctypes.c_short) - 2048
d[np.where(np.abs(d) > 1500)[0]] = 0
return d
Depending on file version, signal is read as data - 2048 for a 1.0 scale factor and 2048 - data for a -1.0 scale factor, or vice versa.
neo reads either as straight data or 4096 - data.
On top of that, even if the values matched, neo doesn't actually take signal inversion into account :
if format_100:
if signal_inv == 1:
read_function = readHDF5t_100
elif signal_inv == 1:
read_function = readHDF5t_100_i
else:
raise Exception("Unknown signal inversion")
else:
if signal_inv == 1:
read_function = readHDF5t_101
elif signal_inv == 1:
read_function = readHDF5t_101_i
So the read function defaults to readHDF5t_101 or readHDF5t_100 every time.
I'm not sure if the way HS2 accesses data shifts values by 2048 which would explain the difference, but at the very least signal inversion should be taken into account. Will submit an issue on neo.