vcfR_to_fasta script introduces NAs instead of Ns creating a frameshift
Hello @PhHermann
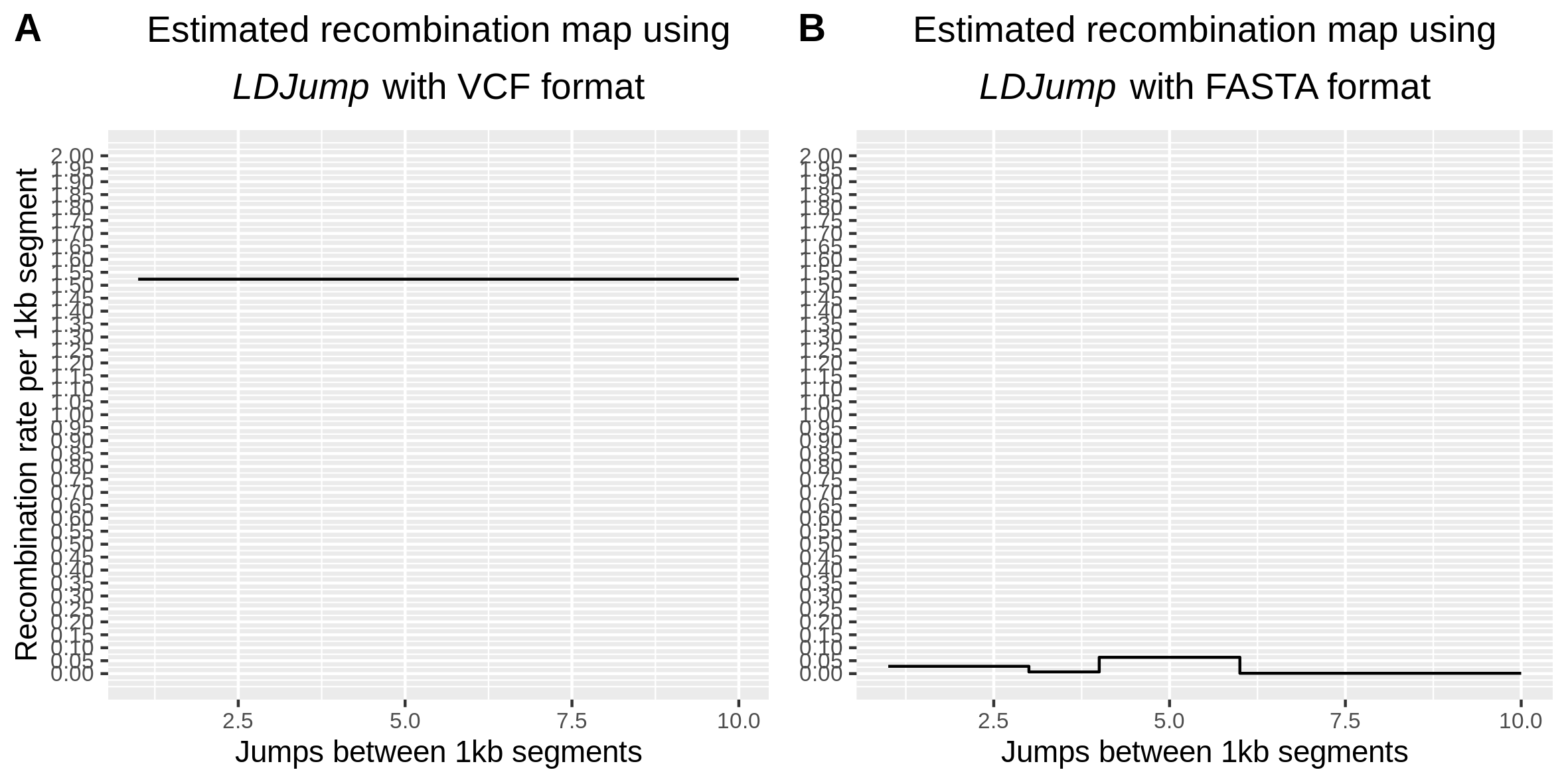
First of all thank you for this great package. I am attempting to run LDJump using a vcf file that contains missing data. vcfR introduces NAs instead of Ns for missing sites, hence it creates a frameshift and the sequences do not appear aligned any more, inflating the estimated recombination rates. I have converted my vcf file to fasta format using GATK FastaAlternateReferenceMaker masking the missing data and then run LDJump (fasta option) ; as you can see in the picture I have attached below, the estimates I am obtaining are very different as expected. (I have also attached the vcfR_to_fasta generated file and the original vcf) Do you think there could be a way to solve this issue? Additionally, missing data cause Phi to crash producing the following error: Floating point exception (core dumped)
Thank you very much in advance, Clio
sel_1_1001.recode.vcf.fasta.txt
Hello @biolevol, I am sorry that there has not been any response for your question. Thank you for taking interest into LDJump and giving a structured question. I checked your produced FASTA file, and it seems that the conversion results in a FASTA file with no variation - this is why LDJump estimates the same recombination rate for the whole sequence.
About your VCF file: What genetic data are you working with, is it from a haploid or diploid organism?
vcfR principally should convert missing positions into ambiguous characters.
What FASTA reference sequence are you using to convert from VCF to FASTA?