Error: have not read haploid/diploid status
Hey @PhHermann
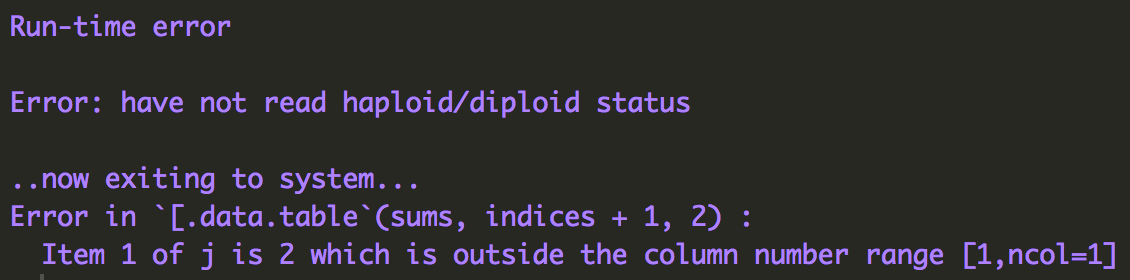
I'm receiving an error when trying to run LDJump on some FASTA files. The error occurs whether I'm running FASTA files included as part of LDJump examples or my own FASTA files. I've included an image of the error below.
Here is the command I ran that generates the error above.
LDJump("/Users/jamessantangelo/Documents/Academia/Doctorate_PhD/Projects/MRT_Mutation-Recombination-Theory/data/test/N100_bot0.9/fasta-files/N100_bot0.9_gen1000_sorted.fasta",
alpha = 0.05, segLength = 100000,
pathLDhat = "/Users/jamessantangelo/downloaded-utilities/LDhat",
pathPhi = "/Users/jamessantangelo/downloaded-utilities/PhiPack/Phi",
format = "fasta", constant = TRUE, status = TRUE, cores = 4,
accept = TRUE, demography = FALSE, out = "test")
It seems the error is happening because LDhat's convert utility requires the FASTA file's first line to be n_seqs L_seqs haploid(1)/diploid(2) but LDJump is not adding this line to the FASTA sent to LDhat. I've tried adding it to the FASTA files myself but then LDJump will no accept them as input.
Any idea how I can solve this problem?
Thanks!
I had the same issue and think it's because "segLength" was set above 100000, which would turn the number into scientific notation for the segment fasta file for LDhat. If I ran it with 10000, I was able to pass that.